Introduction
LINDTIE is a tool designed to identify aberrant transcripts in cancer using long-read RNA-seq data generated by platforms such as Oxford Nanopore Technologies (ONT) and PacBio. It extends beyond canonical gene fusions to capture the full spectrum of cancer transcriptome rearrangements, including fusion, transcribed structural variant (TSV), and novel splice variant (NSV). The pipeline accepts raw transcriptome (RNA-seq) FASTQ files from case and control samples and produces TSV files containing the novel variants it identifies.
LINDTIE uses a hybrid strategy that integrates reference-free de novo assembly with reference-guided assembly, combined with differential transcript expression analysis, to uncover previously uncharacterised transcripts. The workflow consists of four core procedures: assembly, quantification, differential expression analysis, and annotation.
LINDTIE is implemented in Nextflow, providing users with enhanced control over pipeline execution, including the ability to interrupt runs, adjust parameters from the command line, and resume analyses from previous checkpoints.
git clone https://github.com/jiawei-tan/LINDTIE.git & modify nextflow.configcases/ & controls/nextflow runPipeline Overview
LINDTIE employs a one-to-N case–control design and a four-stage analysis workflow to identify aberrant transcripts from long-read RNA-seq data. Understanding this workflow helps you interpret results and troubleshoot issues.
Workflow Diagram
Installation + Configuration
Prerequisites
LINDTIE is built using Nextflow.
Before running the pipeline, ensure that the following are installed or available on your Linux-based system:
- Nextflow
- A container engine: Singularity / Apptainer (recommended for HPCs) or Docker
Many HPC systems provide Nextflow and Singularity as environment modules. If your system uses modules, you can check availability with module avail and load them with a command such as: module load nextflow/<version> singularity/<version>
Installing from GitHub
Clone the LINDTIE repository:
git clone https://github.com/jiawei-tan/LINDTIE.gitConfiguration
Navigate to the LINDTIE base directory to begin configuring the pipeline:
cd LINDTIEEdit the Nextflow configuration file (nextflow.config), located in the LINDTIE base directory:
i. Process Configuration (Executor, Queue, and Resource Profiles)
Under the process block in nextflow.config, you can specify the executor used by your HPC system, the queue to submit jobs to, and resource profiles for different types of tasks.
Select an HPC executor and queue:
Choose an appropriate Nextflow executor (e.g., slurm, pbs, sge, lsf, local, etc.) supported by your compute environment. The default configuration supplied with LINDTIE is optimized for WEHI's Milton HPC, which uses the SLURM workload manager. Refer to the Nextflow documentation for the full list of available executors.
Example (default SLURM configuration):
// Process execution configuration – modify as required
process {
executor = 'slurm'
queue = 'regular' // default SLURM queue
cache = 'lenient'
errorStrategy = 'retry' // default retry failed tasks
}For further details on customizing Nextflow configuration files, see the official documentation.
Resource Profiles with Labels:
LINDTIE assigns resource requirements to tasks using Nextflow labels. The default settings allocate resources appropriate for typical HPC environments, but you may reduce or increase these values depending on your system’s available resources.
Example (default label-specific resource settings):
// Configuration for short-running, lightweight tasks
withLabel: 'process_short' {
cpus = 1
memory = 4.GB // 4 GB RAM
time = 1.h // 1-hour time limit
}
// Configuration for moderately intensive tasks
withLabel: 'process_medium' {
cpus = 8
memory = 16.GB // 16 GB RAM
time = 8.h // 8-hour time limit
}
// Configuration for long-running, resource-heavy tasks
withLabel: 'process_long' {
cpus = 16
memory = 64.GB // 64 GB RAM
time = 16.h // 16-hour time limit
}ii. Environment Configuration (JVM Memory Limits)
Under the env block, you can adjust the Java Virtual Machine (JVM) heap size for Nextflow:
Limit the JVM heap size:
The default setting allocates 100 GB of heap memory, which you may reduce or increase depending on your system’s available resources.
Adjusting this value ensures that Nextflow does not exceed the memory limits enforced by your HPC scheduler.
Example (default JVM heap size):
env {
// JVM heap size (-Xmx sets the maximum heap memory) - default 100GB
NXF_JVM_ARGS = '-Xmx100g'
}Recommended requirements:
- CPUs = 48
- Memory = 100GB
Setting Up References
Download the compressed pre-built reference package from Zenodo (1.06GB):
curl -O https://zenodo.org/records/18531809/files/LINDTIE_ref.tar.gz
# decompress the tar.gz file and remove the tar.gz file
tar xzf LINDTIE_ref.tar.gz && rm LINDTIE_ref.tar.gzThis will generate a ref directory containing the seven required reference files. Ensure that the ref directory is placed inside the LINDTIE base directory:
LINDTIE/
└── ref/
├── chess3.0_with_HTLV1_HPV_HBV_HIV1_HIV2_EBV.fa
├── chess3.0_with_HTLV1_HPV_HBV_HIV1_HIV2_EBV.gtf
├── chess3.0_with_HTLV1_HPV_HBV_HIV1_HIV2_EBV.info
├── Cosmic_CancerGeneCensus_v103_GRCh38_tier_fusion.tsv
├── hg38_splice_junctions.bed
├── hg38_with_HTLV1_HPV_HBV_HIV1_HIV2_EBV.fa
└── tx2gene.txt
The reference comprises both human and viral sequences, including viruses known to integrate into the host genome, such as HTLV-1 (NC_001436.1), HPV (NC_027779.1), HBV (NC_003977.2), HIV-1 (NC_001802.1), HIV-2 (NC_001722.1), and EBV (NC_009334.1).
Running LINDTIE
In the directory where you will run LINDTIE, create the required cases and controls subdirectories:
mkdir -p cases
mkdir -p controlsInput Files
Allocate the long-read RNA-seq data in FASTQ or FASTA format (can be gzipped) into the appropriate directories for your case and control samples.
Cases refer to the cancer samples in which you want to identify variants, while controls are used as the reference for comparison. Ideally, control samples should be benign tissue of the same type as the primary tumour. If this is not feasible, such as in blood cancers, acceptable alternatives include remission samples or samples from other individuals, ideally of the same cancer type.
Including more controls increases statistical power; aim for a minimum of 2, with 10 to 15 being optimal.
Run LINDTIE with Nextflow
Run LINDTIE with one of the following commands:
nextflow run LINDTIE/main.nf -params-file LINDTIE/params.yaml -profile singularitynextflow run LINDTIE/main.nf -params-file LINDTIE/params.yaml -profile dockerChoose the appropriate value for -profile based on the container engine supported by your system (singularity or docker).
You can also run nextflow run LINDTIE/main.nf --help to see all available options and parameters.
Submitting the Nextflow Script as a Job
A more effective approach than launching the Nextflow driver job from the login node is to wrap the Nextflow run command in a script and submit the workflow as a job.
A run_LINDTIE.sh template bash script is provided in the LINDTIE base directory:
#!/bin/bash
#SBATCH --job-name=run_LINDTIE
#SBATCH --partition=regular
#SBATCH --ntasks=1
#SBATCH --ntasks-per-node=1
#SBATCH --cpus-per-task=1
#SBATCH --mem=8G
#SBATCH --time=24:00:00
#SBATCH --mail-type=BEGIN,END,FAIL
#SBATCH --mail-user=your-email@example.com
#SBATCH --output=script_output/%x_%J.out
#SBATCH --error=script_output/%x_%J.err
module load nextflow/25.04.2 singularity/4.1.5
# modify the path to the LINDTIE base directory
LINDTIE_dir=/path/to/your/LINDTIE
nextflow run $LINDTIE_dir/main.nf -params-file $LINDTIE_dir/params.yaml -profile singularityFine Tuning Parameters
Adjust these parameters based on your sample characteristics and analysis requirements. Parameters marked with ⭐ are commonly modified.
| Parameter | Description | Default | Options/Format | Category |
|---|---|---|---|---|
⭐ assembly_mode |
Determines the assembly strategy used by the pipeline.
|
hybrid |
"hybrid", "denovo", "denovo_subset", or "ref_guided" |
Assembly |
⭐ rnabloom2_preset |
Sequencing platform preset for RNA-Bloom2 assembly | (empty) | (empty) or "lrpb" |
Assembly |
⭐ minimap2_preset |
Preset configuration for Minimap2 alignment (passed to -ax) |
map-ont |
"map-ont", "map-pb", "map-hifi", or "lr:hq" |
Quantification |
⭐ subset_count |
Specifies the number of reads to subset when the "denovo_subset" mode is selected. |
NULL |
integer (e.g., 1000000) or NULL |
Assembly |
oarfish_num_bootstraps |
Number of bootstrap iterations for quantification uncertainty | 10 |
integer (e.g., 10) |
Quantification |
RUN_DE |
Toggle to enable/disable the Differential Expression module | true |
true or false |
DE Analysis |
fdr |
FDR significance threshold for differentially expressed genes | 0.05 |
numeric (e.g., 0.05) |
DE Analysis |
min_cpm |
Minimum CPM required for a gene to be considered expressed | 0.5 |
numeric (e.g., 0.5) |
DE Analysis |
min_logfc |
Minimum absolute Log2 Fold Change for DE detection | 2 |
numeric (e.g., 2) |
DE Analysis |
⭐detect_viral_integration |
Toggle to enable or disable the detection of viral integration variants | false |
true or false |
Detection |
min_clip |
Minimum clipped sequence length to trigger SV detection | 20 |
integer (e.g., 20) |
Detection |
min_gap |
Minimum gap size between aligned segments for events | 7 |
integer (e.g., 7) |
Detection |
min_match |
Sequence matching quality thresholds (length,identity) | 30,0.3 |
string (e.g., "30,0.3") |
Detection |
splice_motif_mismatch |
Maximum allowed mismatches for splice motifs | 1 |
integer (e.g., 1) |
Detection |
single_sample_min_vaf |
Minimum variant allele frequency (VAF) threshold for retaining variants when RUN_DE is false |
0.1 |
numeric (e.g., 0.1) |
Detection |
gene_filter |
Whitelist of specific gene symbols to include | NULL |
comma-separated or NULL (e.g., "TP53,BRCA2") |
Filtering |
var_filter |
Whitelist of variant types to include | NULL |
comma-separated or NULL (e.g., "DEL,INS") |
Filtering |
Configuration Methods
There are two ways to configure the parameters for LINDTIE:
Option 1: Edit the params.yaml file before running
- Open the
params.yamlfile located in theLINDTIEbase directory. - Modify any parameters as needed.
- Save the file and then run LINDTIE.
Option 2: Override parameters at runtime using command line arguments
- Run LINDTIE with the desired parameters using the command line.
# Default parameters for the workflow
# Assembly Mode: 'hybrid', 'denovo', 'denovo_subset', or 'ref_guided'
assembly_mode: 'hybrid'
# Tool Presets
# minimap2 presets (passed to -ax):
# 'map-ont' : Oxford Nanopore genomic reads (default)
# 'map-pb' : PacBio CLR genomic reads
# 'map-hifi': PacBio HiFi/CCS genomic reads (v2.19+)
# 'lr:hq' : Nanopore Q20 genomic reads (v2.27+)
minimap2_preset: 'map-ont'
# rnabloom2 presets:
# '' : Leave empty for ONT (default)
# '-lrpb' : For PacBio
rnabloom2_preset: ''
subset_count: null # NULL for no subsetting, otherwise the number of reads to subset to
detect_viral_integration: false # true or false (default: false)
RUN_DE: true # true or false (default: true)
fdr: 0.05 # default 0.05
min_cpm: 0.5 # default 0.5
min_logfc: 2 # default 2
min_clip: 20 # default 20
min_gap: 7 # default 7
min_match: '30,0.3' # default '30,0.3'
splice_motif_mismatch: 1 # default 1
oarfish_num_bootstraps: 10 # default 10
gene_filter: NULL # default NULL (e.g. "TP53,BRCA2")
var_filter: NULL # default NULL (e.g. "DEL,INS")
single_sample_min_vaf: 0.1 # default 0.1
nextflow run LINDTIE/main.nf \
-params-file LINDTIE/params.yaml \
-profile singularity \
--rnabloom2_preset "lrpb" \
--minimap2_preset "map-pb" \
--assembly_mode "denovo"For Option 2, use a single dash (-) for Nextflow runtime options; use a double dash (--) for pipeline parameters.
Output
After running LINDTIE, a results directory (<caseName>_output) will be created. This directory is organized by analysis steps, with the final results for the sample stored in the FinalOutput folder:
<caseName>_output/
├── 01-Assembly
├── 02-Quantification
├── 03-DifferentialExpression
├── 04-Annotation
├── FinalOutput
└── run_parameters.logFinalOutput Results
The final results produced by LINDTIE are located in <caseName>_output/FinalOutput/, which contains the following files:
<caseName>_output/FinalOutput/
├── log
├── refined_annotated_contigs.bam
├── refined_annotated_contigs.bam.bai
├── refined_annotated_contigs.fasta
├── refined_annotated_contigs.vcf
├── <caseName>_all_variants_ranked_results.tsv
├── <caseName>_discarded_results.tsv
└── <caseName>_results.tsvPrimary Results: <caseName>_results.tsv
This file is the primary result table. Variants with multiple annotations are collapsed, meaning each contig appears as a single row with consolidated information.
Output File Column Descriptions
| Column # | Column name | Description |
|---|---|---|
| 1 | chr1 | Chromosome for end 1 of the variant. |
| 2 | pos1 | Genomic position for end 1 of the variant. |
| 3 | strand1 | Strand (+/–) for end 1 of the variant. |
| 4 | chr2 | Chromosome for end 2 of the variant. |
| 5 | pos2 | Genomic position for end 2 of the variant. |
| 6 | strand2 | Strand (+/–) for end 2 of the variant. |
| 7 | variant_type | LINDTIE's estimated classification of the variant type. Refer to LINDTIE's Variant Classification for the full list of variant types. |
| 8 | other_variant_type | Consolidated variant annotations for the contig. Multiple types are separated by "|" |
| 9 | overlapping_genes | Genes overlapped by the contig. If separated by colons (":"), each gene corresponds to a different soft/hard-clipped segment. |
| 10 | sample | Sample to which this variant belongs. |
| 11 | variant_id | Assigned variant ID (matches the VCF file). |
| 12 | partner_id | For fusions or junctions with two breakpoints, this identifies the paired variant. |
| 13 | vars_in_contig | Number of variants detected on the aligned contig. |
| 14 | varsize | Size of the variant on the reference genome. |
| 15 | contig_varsize | Size of the variant on the contig sequence. |
| 16 | cpos | Position of the variant on the contig (independent of alignment direction). |
| 17 | TPM | Length-corrected transcript-per-million estimate for the variant contig. |
| 18 | mean_WT_TPM | Mean length-corrected TPM of all wild-type genes associated with this transcript. |
| 19 | VAF | Approximate variant allele frequency estimate. |
| 20 | logFC | Maximum log fold change of associated transcript(s) in the case sample vs. controls. |
| 21 | FDR | Adjusted (multiple-testing corrected) p-value. |
| 22 | PValue | Minimum p-value for transcripts associated with this variant vs. controls. |
| 23 | num_reads_case | Total read counts for all transcripts associated with the contig in the case sample. |
| 24 | total_num_reads_controls | Total read counts for all associated transcripts across control samples. |
| 25 | large_varsize | Indicates whether the variant size exceeds the min_clip threshold (default: 30 bp). |
| 26 | is_contig_spliced | Indicates whether the contig is spliced (i.e., contains alignment gaps). |
| 27 | spliced_exon | Indicates a novel or extended exon variant with a corresponding junction. |
| 28 | overlaps_exon | Indicates whether the variant overlaps any annotated reference exon. |
| 29 | overlaps_gene | Indicates whether the variant overlaps any annotated reference gene. |
| 30 | motif | Splice motif sequence. |
| 31 | valid_motif | Indicates whether the variant contains a valid splice motif. Some variant types (e.g., TSVs or splice events at known boundaries) are not tested. |
| 32 | COSMIC_tier | The Cancer Gene Census assessment for the genes involved. Tier 1 denotes genes with documented activity relevant to cancer; Tier 2 denotes genes with strong evidence of a role in cancer but less extensive documentation. |
| 33 | COSMIC_fusion | Indicates whether any gene involved in the event is listed in COSMIC Fusion. Yes means at least one overlapping gene is reported as a fusion partner in COSMIC; No means none are listed. |
| 34 | site1_feature | The genomic feature annotation at the exact position of end 1 (e.g., CDS, UTR, intron, intergenic). |
| 35 | site2_feature | The genomic feature annotation at the exact position of end 2 (e.g., CDS, UTR, intron, intergenic). |
| 36 | is_coding | Indicates whether the variant involves coding regions. This is determined by evaluating whether site1_feature and site2_feature are annotated as coding sequences (i.e., CDS). |
| 37 | contig_id | Contig name from the de novo assembly. |
| 38 | unique_contig_ID | ID of the modified SuperTranscript used in visualization outputs. |
| 39 | contig_len | Length of the contig sequence. |
| 40 | contig_cigar | CIGAR string representing the contig's genome alignment (may contain two strings if soft/hard-clipped). |
| 41 | seq_loc1 | Location string for the first sequence region (e.g., contig123:100–140). |
| 42 | seq_loc2 | Location string for the second sequence region, if applicable. |
| 43 | seq1 | 20 bp sequence around the main variant site. |
| 44 | seq2 | 20 bp sequence around the second variant site (if applicable). |
| 45 | variant_score | Score used for variant prioritization. |
<caseName>_all_variants_ranked_results.tsv
This file is an expanded version of the <caseName>_results.tsv file. This table lists all variant annotations individually, without collapsing (i.e., a contig may appear in multiple rows if it has multiple annotations). It includes all columns present in <caseName>_results.tsv, plus three additional columns:
| Column # | Column name | Description |
|---|---|---|
| 46 | rank_within_contig | Rank of each annotation for a given contig based on its score; 1 = highest-scoring annotation. |
| 47 | is_primary | Indicates whether this annotation was selected as the primary variant for that contig (i.e., the entry included in <caseName>_results.tsv). |
The other_variant_type column (col 8) is empty in this file because each annotation is shown separately rather than consolidated.
<caseName>_discarded_results.tsv
This file contains variants filtered out due to low-complexity sequences. Specifically, variants are placed in this table if the seq1 or seq2 columns contain:
- a polyA or polyT stretch of ≥ 10 bp, or
- a perfect dinucleotide repeat of ≥ 10 repeats (i.e., 20 bp total).
Visualization Files
The following files are useful for inspection in IGV to visualize alignments and examine the refined contig sequences:
refined_annotated_contigs.bam/.bam.bai: BAM and BAM index files that contain the aligned refined transcript sequences for visualization.refined_annotated_contigs.fasta: A FASTA file that contains the refined transcript sequences used in the analysis.refined_annotated_contigs.vcf: A VCF file that lists the refined variant calls.
Intermediate Files Generated at Each Step
LINDTIE produces several intermediate files throughout the pipeline. These files can be useful for troubleshooting, quality checks, or deeper inspection of specific steps.
<caseName>_output/
├── run_parameters.log
├── 01-Assembly
│ ├── denovo_read_counts.log (assembly_mode = hybrid or denovo or denovo_subset)
│ ├── rnabloom.transcripts.fa (assembly_mode = hybrid or denovo or denovo_subset)
│ ├── read_counts_summary.log (assembly_mode = hybrid or ref_guided or denovo_subset)
│ ├── confident_mapped.bam & confident_mapped.bam.bai (assembly_mode = hybrid or ref_guided or denovo_subset)
│ ├── reads_all_sorted.bam & reads_all_sorted.bam.bai (assembly_mode = hybrid or ref_guided or denovo_subset)
│ ├── stringtie2_assembly.fa (assembly_mode = hybrid or ref_guided or denovo_subset)
│ └── stringtie2_assembly.gtf (assembly_mode = hybrid or ref_guided or denovo_subset)
├── 02-Quantification
│ ├── cases
│ │ ├── <caseName>.infreps.pq
│ │ ├── <caseName>.meta_info.json
│ │ └── <caseName>.quant
│ └── controls
│ ├── <controlName>.infreps.pq
│ ├── <controlName>.meta_info.json
│ └── <controlName>.quant
├── 03-DifferentialExpression
│ ├── DE_contigs.fasta
│ ├── DE_contigs_mapped_to_hg38.bam
│ ├── DE_contigs_mapped_to_hg38.bam.bai
│ ├── DE.log
│ ├── DE_MD_plot.png
│ ├── DE_MDS_plot.png
│ ├── DE_QLDisp_plot.png
│ ├── DE_transcript_full_results.txt
│ └── DE_transcript_significant.txt
└── 04-Annotation
├── annotated_contigs.bam
├── annotated_contigs.bam.bai
├── annotated_contigs_info.tsv
├── annotated_contigs.vcf
└── annotation.logrun_parameters.log
A log file that contains the run parameters used to run LINDTIE.
01-Assembly
denovo_read_counts.log: A log file that contains the read counts for the de novo assembly. Only present when assembly_mode = denovo or denovo_subset.rnabloom.transcripts.fa: A FASTA file that contains the assembled transcript sequences produced by RNA-Bloom2. Only present when assembly_mode = hybrid or denovo or denovo_subset.read_counts_summary.log: A log file that contains the read counts summary. Only present when assembly_mode = hybrid or ref_guided or denovo_subset.confident_mapped.bam: A BAM file that contains the confident mapped reads. Only present when assembly_mode = hybrid or ref_guided or denovo_subset.reads_all_sorted.bam: A BAM file that contains all the reads mapped to the reference genome. Only present when assembly_mode = hybrid or ref_guided or denovo_subset.stringtie2_assembly.fa: A FASTA file that contains the assembled transcript sequences produced by StringTie2. Only present when assembly_mode = hybrid or ref_guided or denovo_subset.stringtie2_assembly.gtf: A GTF file that contains the assembled transcript annotations produced by StringTie2. Only present when assembly_mode = hybrid or ref_guided or denovo_subset.read_counts_summary.log: A log file that contains the read counts summary. Only present when assembly_mode = hybrid or ref_guided or denovo_subset.
02-Quantification
Files generated by Oarfish for both the case and control samples:
<sampleName>.quant: A tab-separated file that contains the quantified transcripts along with their lengths, metadata, and the estimated number of reads originating from each transcript.<sampleName>.meta_info.json: A JSON file that contains the parameters used to run Oarfish and other sample-level metadata excluding transcript quantifications.<sampleName>.infreps.pq: A Parquet file that contains estimated transcript counts, with each row representing a transcript and each column representing an inferential replicate.
03-DifferentialExpression
Files containing results from differential expression analysis of assembled transcripts:
DE_contigs.fasta: A FASTA file that contains the transcripts sequences identified as significantly differentially expressed.DE_contigs_mapped_to_hg38.bam/DE_contigs_mapped_to_hg38.bam.bai: BAM and BAM index files that contain the alignments of differentially expressed transcripts to the hg38 reference genome for visualization.DE.log: A log file that contains the log messages from the differential expression analysis.DE_MD_plot.png: A PNG file that contains the mean–difference (MD) plot of expression changes.DE_MDS_plot.png: A PNG file that contains the multidimensional scaling (MDS) plot showing sample relationships.DE_QLDisp_plot.png: A PNG file that contains the quasi-likelihood dispersion diagnostic plot.DE_transcript_full_results.txt: A text file that contains the complete statistical results for all transcripts tested.DE_transcript_significant.txt: A text file that contains the subset of transcripts identified as significantly differentially expressed.
04-Annotation
Files containing structural and functional annotations of transcripts:
annotated_contigs.bam/annotated_contigs.bam.bai: BAM and BAM index files that contain the annotated transcript alignments generated from alignment-based analysis.annotated_contigs_info.tsv: A tab-separated file that contains metadata and functional annotations for each transcript.annotated_contigs.vcf: A VCF file that contains variant calls identified during the annotation process.annotation.log: A log file that contains log messages from the annotation workflow.
LINDTIE's Variant Classification
The following are the variant types identified and classified by LINDTIE:
| Variant Type | Full Name | Description | Condition |
|---|---|---|---|
| FUS | Fusion | Inter-chromosomal or distant intra-chromosomal rearrangement | When two reads from the same contig map to different genomic locations with clipping events |
| IGR | Intra-Genic Rearrangement | Rearrangement within the same gene | When both parts of a fusion occur within the same gene(s) |
| UN | Unknown | Soft-clipped sequence of unknown origin | When soft-clipped sequence is present but not part of a fusion event |
| INS | Insertion | Sequence inserted relative to the reference | When CIGAR contains an insertion operation with size ≥ MIN_GAP |
| DEL | Deletion | Sequence deleted relative to the reference | When CIGAR contains a deletion operation with size ≥ MIN_GAP |
| EE | Extended Exon | Extension of known exonic sequence | When a novel block extends beyond existing exon boundaries (either left or right side) |
| NE | Novel Exon | Completely novel exonic sequence | When a novel block doesn't overlap any known exonic regions |
| RI | Retained Intron | Intronic sequence retained in the transcript | When a novel block spans between two exons (has both left and right exonic boundaries) |
| AS | Alternative Splicing | Alternative splicing event using known splice sites | When both ends of a junction match known splice sites but the combination is novel |
| NEJ | Novel Exon Junction | Completely novel splice junction | When neither end of a novel junction matches known splice sites |
| PNJ | Partial Novel Junction | Junction with one known and one novel splice site | When only one end of a novel junction matches known splice sites |
LINDTIE's variant classification rules and filtering logic are based on the following criteria:
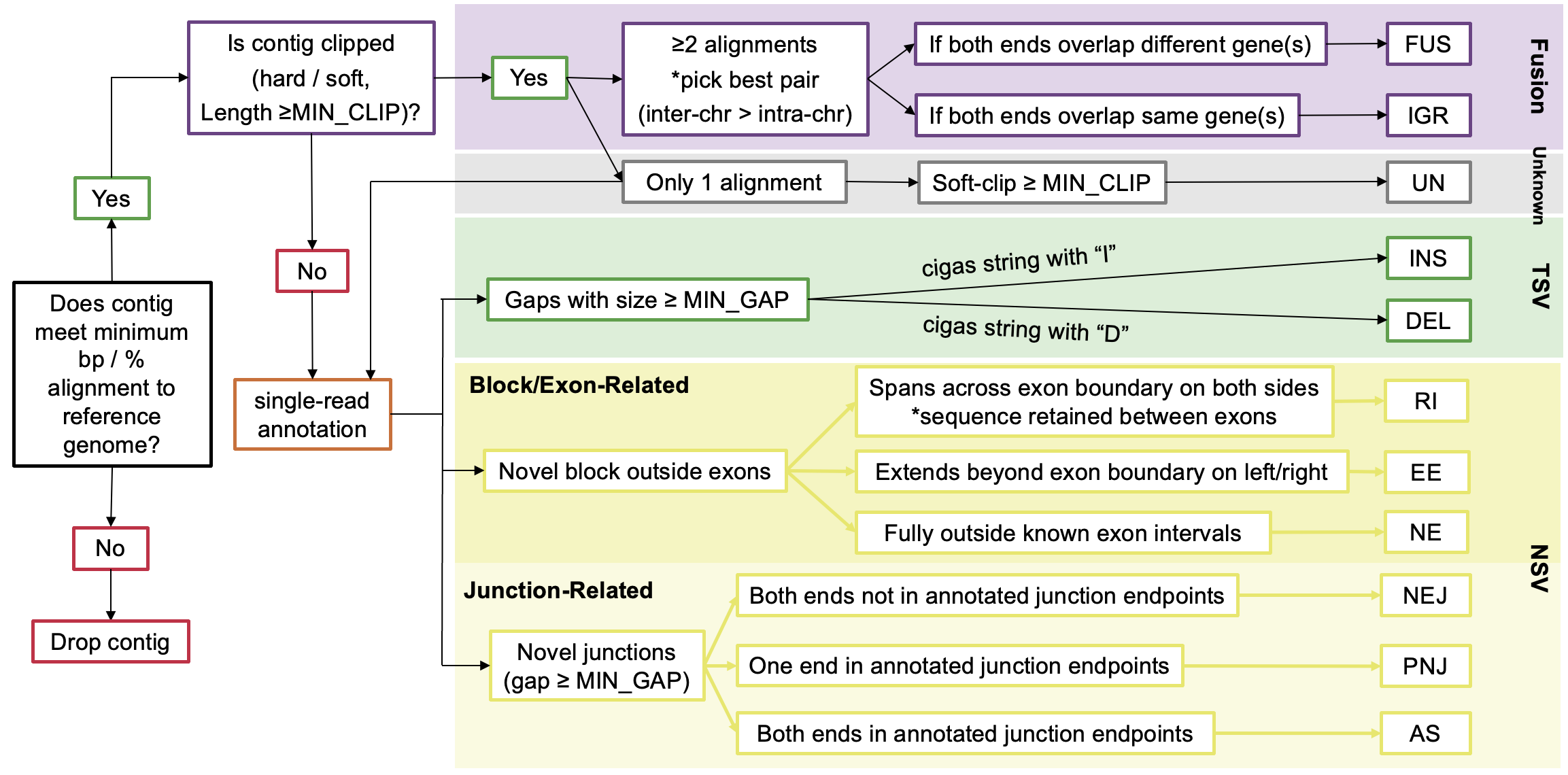
LINDTIE's Variant-Specific Criteria
The following are the variant-specific criteria used by LINDTIE to classify the variants:
| Variant Type | Full name | Category | Clipping | Spliced Contig1 | Variant Size | Overlaps Gene | Overlaps Exon | Spliced Exon2 | Valid Motif3 |
|---|---|---|---|---|---|---|---|---|---|
| FUS | Fusion | Fusion | hard/soft | - | >min_clip | ✅️ | - | - | - |
| IGR | Intra-Genic Rearrangement | Fusion | hard/soft | - | >min_clip | ✅️ | - | - | - |
| UN | Unknown | Unknown | soft | - | >min_clip | ✅️ | - | - | - |
| INS | Insertion | TSV | - | ✅️ | >min_gap | ✅️ | ✅️ | - | - |
| DEL | Deletion | TSV | - | ✅️ | >min_gap | ✅️ | ✅️ | - | - |
| RI | Retained Intron | NSV | - | ✅️ | >min_clip | ✅️ | ✅️ | - | - |
| EE | Extended Exon | NSV | - | ✅️ | >min_clip | ✅️ | ❌ | ✅️ | ✅️ |
| NE | Novel Exon | NSV | - | ✅️ | >min_clip | ❌ | ❌ | ✅️ | - |
| NEJ | Novel Exon Junction | NSV | - | ✅️ | >min_gap | ✅️ | ✅️ | ✅️ | ✅️ |
| PNJ | Partial Novel Junction | NSV | - | ✅️ | >min_gap | ✅️ | ✅️ | ✅️ | ✅️ |
| AS | Alternative Splicing | NSV | - | ✅️ | >min_gap | ✅️ | ✅️ | - | - |
[1] Spliced Contig: Any alignment containing a splice (≥1 gap)
[2] Spliced Exon: EE/NE variants that have adjacent supporting junctions, and for selected junction variants themselves.
[3] Valid Motif: True when the 2-bp splice motifs at the relevant boundaries match canonical GT-AG (or CT-AC on the opposite strand) within the allowed mismatch tolerance
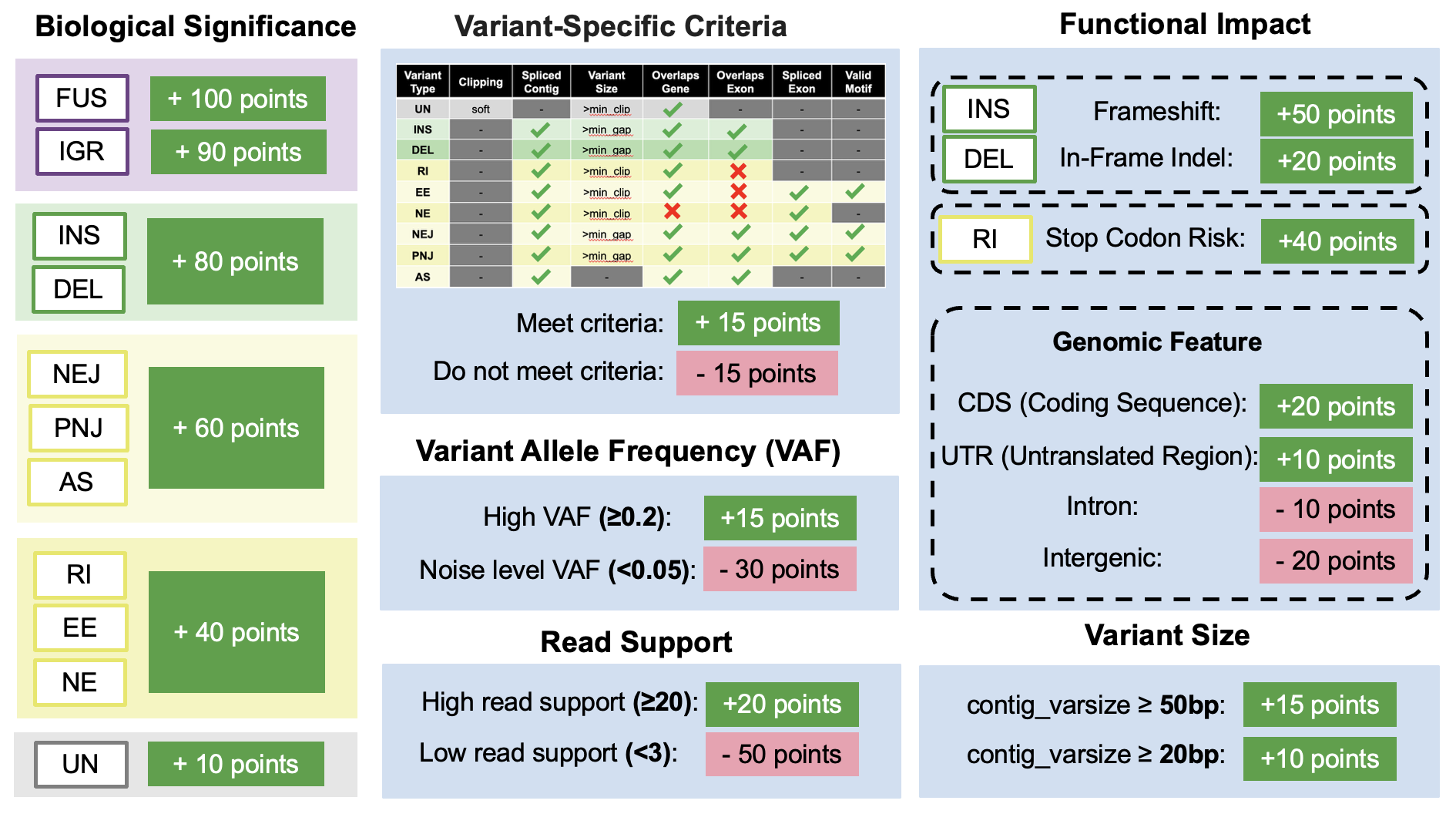
LINDTIE’s Scoring System
LINDTIE's scoring system is based on the following criteria:
Resume Your Run
You can easily resume your run in case of changes to the parameters or inputs using -resume. Nextflow will try to not recalculate steps that are already done:
nextflow run LINDTIE/main.nf -params-file LINDTIE/params.yaml -resumeOnly a single dash (-) is needed for the resume flag.
Nextflow will need access to the working directory where temporary calculations are stored. Per default, this is set to work but can be adjusted via -w /path/to/any/workdir. In addition, the .nextflow.log file is needed to resume a run, thus, this will only work if you resume the run from the same folder where you started it.
Testing LINDTIE
Example test data is included to help you quickly verify that the pipeline is running correctly. The test set contains one case sample and two control samples, located in the test_case directory under the LINDTIE base directory:
LINDTIE/
└──test_case/
├── cases
│ └── test-case.fastq.gz
├── controls
│ ├── test-control0.fastq.gz
│ └── test-control1.fastq.gz
└── run_LINDTIE.sh
You can test LINDTIE either by running the following command directly in the terminal or by executing the provided run_LINDTIE.sh script (make sure to modify the path):
# modify the path to the LINDTIE base directory
LINDTIE_dir=/path/to/your/LINDTIE
nextflow run $LINDTIE_dir/main.nf -params-file $LINDTIE_dir/params.yaml -profile singularity
cases/*.fastq.gz controls/*.fastq.gzApproximate run time: 8-10 minutes
Once LINDTIE has finished running, you should see output on the terminal similar to the following:
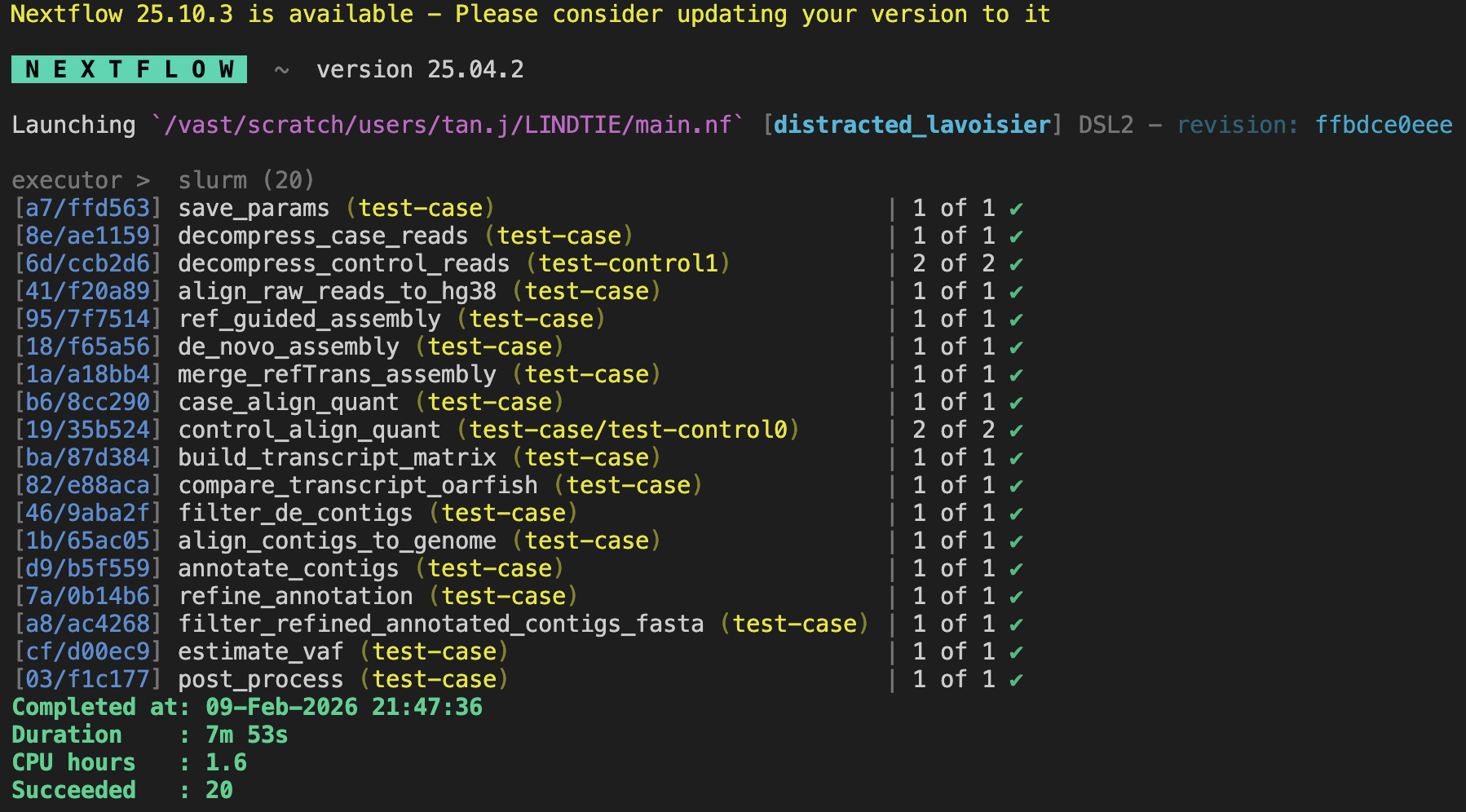
View the collapsed results at: test-case_output/FinalOutput/test-case_results.tsv
The table below summarizes the number of variants detected for each variant type in the test case:
| Variant Type | Count |
|---|---|
| AS | 5 |
| DEL | 6 |
| EE | 6 |
| FUS | 15 |
| IGR | 1 |
| INS | 15 |
| NE | 1 |
| NEJ | 4 |
| PNJ | 9 |
| RI | 4 |
| UN | 4 |
| TOTAL | 70 |
The exact counts may vary between runs. The de novo assembly step (RNA-Bloom2) is not fully deterministic, so the assembled transcripts and therefore the final variants detected may differ slightly each time. However, the overall results should remain broadly consistent.
Examples of Use
Detailed examples and use cases are currently being prepared. Check back soon for comprehensive tutorials and real-world applications of LINDTIE.
Best Practices
Best practices will be added in future releases. Please check back for updates or visit the GitHub repository for the latest information.
Full List of Tools Used in LINDTIE
Listed below are all software tools and version numbers packaged within the Nextflow container used by LINDTIE:
bioconda::rnabloom=2.0.1
bioconda::gffread=0.12.7
bioconda::stringtie=2.2.3
bioconda::minimap2=2.30
bioconda::samtools=1.22
bioconda::bbmap=39.52
bioconda::oarfish=0.8.1
bioconda::bio=1.8.0
bioconda::pysam=0.23.3
bioconda::pybedtools=0.12.0
bioconda::bioconductor-edger=4.4.0
bioconda::bioconductor-tximport=1.34.0
conda-forge::pandas=2.3.0
conda-forge::numpy=2.3.0
conda-forge::intervaltree=3.1.0
conda-forge::r-dplyr=1.1.4
conda-forge::r-data.table=1.17.6
conda-forge::r-jsonlite=2.0.0
conda-forge::r-readr=2.1.5
conda-forge::r-arrow=19.0.1Glossary of Terms
This glossary defines key terms used throughout the LINDTIE documentation and output files.
General Terms
- Aberrant Transcript
- A transcript that differs from the normal reference transcriptome, potentially caused by genomic rearrangements, novel splicing, or other alterations. In cancer, aberrant transcripts may drive tumor growth or serve as biomarkers.
- Contig
- A contiguous sequence assembled from overlapping reads. In LINDTIE, contigs represent assembled transcript sequences that are then analyzed for variants.
- De Novo Assembly
- The process of assembling reads into contigs without using a reference genome. This approach can detect novel sequences not present in the reference.
- Fusion Transcript
- A chimeric RNA molecule containing sequences from two different genes, typically resulting from chromosomal rearrangements. Examples include BCR-ABL in CML and EML4-ALK in lung cancer.
- Long-read RNA-seq (lrRNA-seq)
- RNA sequencing using technologies that produce reads thousands of bases long (ONT, PacBio), enabling full-length transcript sequencing and better detection of structural variants.
- Splice Motif
- The conserved sequence at splice junctions. The canonical splice motif is GT-AG (GT at the 5' donor site, AG at the 3' acceptor site). LINDTIE validates splice junctions against known motifs.
Statistical Terms
- CPM (Counts Per Million)
- A normalization method that scales raw read counts to per-million reads, allowing comparison between samples with different sequencing depths. Formula: CPM = (read count / total reads) × 1,000,000
- FDR (False Discovery Rate)
- The expected proportion of false positives among all significant results. An FDR of 0.05 means 5% of significant results are expected to be false positives. Lower FDR = higher confidence.
- logFC (Log Fold Change)
- The log2-transformed ratio of expression between case and control samples. A logFC of 2 means 4x higher expression in cases; logFC of -2 means 4x lower expression in cases.
- P-value
- The probability of observing the data (or more extreme) if there is no true difference between groups. Lower p-values indicate stronger evidence against the null hypothesis.
- TPM (Transcripts Per Million)
- A normalization method that accounts for both sequencing depth and transcript length, making values comparable across samples and genes. More appropriate than CPM for comparing expression levels of different transcripts.
- VAF (Variant Allele Frequency)
- The proportion of reads supporting the variant allele versus the total reads at that position. Higher VAF suggests the variant is present in more cells (clonal) rather than a subclonal event.
Output Field Terms
- CIGAR String
- A compact representation of how a sequence aligns to a reference. Characters include: M (match/mismatch), I (insertion), D (deletion), N (skipped region/intron), S (soft clip). Example: "100M50N100M" = 100 bases match, 50 base intron, 100 bases match.
Benchmarking & Performance
Benchmarking data will be added in future releases. Please check back for updates or visit the GitHub repository for the latest information.
Troubleshooting
If you encounter issues not covered here, please:
- Check the GitHub issues page
- Review the Nextflow documentation
- Open a new issue on GitHub with detailed information about your problem
Common troubleshooting tips and solutions will be added as they are identified by the community.
Frequently Asked Questions
This section will be populated with frequently asked questions as they arise from the community. In the meantime, please refer to the documentation sections or open an issue on GitHub for specific questions.
Changelog
This page documents all notable changes to LINDTIE. The format is based on Keep a Changelog, and this project adheres to Semantic Versioning.
v0.1.0 - Initial Release
Released: 2025
Added
- Initial release of LINDTIE pipeline
- De novo assembly using RNA-Bloom for long-read data
- Quantification with Oarfish and minimap2 alignment
- Differential expression analysis between case and control samples
- Comprehensive variant annotation system
- Support for Oxford Nanopore Technologies (ONT) data
- Support for PacBio long-read data
- Nextflow-based workflow for HPC environments
- Docker and Singularity container support
- Configurable resource profiles (short, medium, long processes)
- TSV output with ranked variant results
Tool Versions
- RNA-Bloom2: 2.0.1
- minimap2: 2.30
- samtools: 1.22
- Oarfish: 0.8.1
- pandas: 2.3.0
- bio: 1.8.0
- pysam: 0.23.3
- pybedtools: 0.12.0
- edgeR: 4.4.0
- tximport: 1.34.0
- numpy: 2.3.0
- intervaltree: 3.1.0
- dplyr: 1.1.4
- data.table: 1.17.6
- jsonlite: 2.0.0
- readr: 2.1.5
- arrow: 19.0.1
When upgrading between versions, we recommend:
- Review the changelog for breaking changes
- Back up your current configuration files
- Pull the latest version from GitHub
- Re-run with a test dataset to verify functionality
Version Policy
LINDTIE follows semantic versioning (MAJOR.MINOR.PATCH):
- MAJOR: Incompatible changes to input/output format or parameters
- MINOR: New features added in a backward-compatible manner
- PATCH: Bug fixes and minor improvements
Migration Guides
Migration guides will be provided here when new major versions are released. Each guide will detail any breaking changes and provide step-by-step instructions for updating your workflow.
Citing LINDTIE
If you use LINDTIE in your research, please cite:
Citation information will be provided upon publication. Please check the GitHub repository or contact the authors for the most current citation information.